Chapter 8 CosMx
CosMx Spatial Molecular Imager (SMI) from NanoString Technologies (Now Bruker Corporation) is a cutting-edge single-cell resolution spatial transcriptomics platform. Similary to 10x Genomics Xenium platform, CosMx offers subcellular-resolution imaging with multi‑omic detection, allowing researchers to capture up to 6,000 RNA targets and over 64 protein simultaneously. Moreover, it is fully compatible with formalin-fixed paraffin-embedded (FFPE) or fresh-frozen (FF) tissues, which can be applied to both basic (e.g., mouse, cell lines, etc.) and translational research (e.g., clinical samples).
Since CosMx is an imaging-based platform, it does not require sequencing or additional processing of the sequences. The general workflow of a CosMx experiment has been illustrated below:
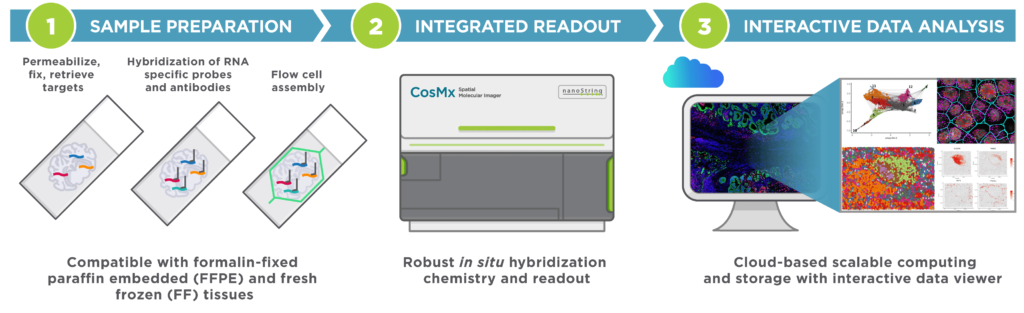
CosMx workflow: The whole workflow contains sample preparation, CosMx in situ processing, and downstream analysis. (Image from: https://nanostring.com/products/cosmx-spatial-molecular-imager/single-cell-imaging-overview//)
8.1 Input preparation
Here, we use public a dataset provided by NanoString to demonstrate the pipeline.
This human frontal cortex FFPE data is available through: https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/human-frontal-cortex-ffpe-dataset/. It is the first single-cell level spatial transcriptomics dataset generated by CosMx with 6,000 genes detected. Users can download the Basic Data Files through the Download Data tab. The basic data files are ~15Gb in total.
In the following example, I will use two datasets, S3 and S4 to run SpaceSequest. If you don’t have S3, simply copy S3 to S4 following by renaming the files would work.
~/SpaceSequest_demo/5_CosMx/
├── S3/
├── S3_exprMat_file.csv
├── S3_fov_positions_file.csv
├── S3_metadata_file.csv
├── S3-polygons.csv
└── S3_tx_file.csv
├── S4/
├── S4_exprMat_file.csv
├── S4_fov_positions_file.csv
├── S4_metadata_file.csv
├── S4-polygons.csv
└── S4_tx_file.csv8.2 Pipeline setup
Demo run directory: ~/SpaceSequest_demo/5_CosMx
First, we initiate the pipeline using the following script. This will generate templates of config.yml and sampleMeta.csv:
#First step, generate config and sampleMeta file
cosmx ~/SpaceSequest_demo/5_CosMxThen, fill in the config.yml file and the sampleMeta.csv file as below:
#config file for CosMx. Please avoid using spaces in names or paths. All items are required.
project_ID: CoxMx_demo #required project name
sampleMeta: ~/SpaceSequest_demo/5_CosMx/sampleMeta.csv #path to the sampleMeta file
output_dir: ~/SpaceSequest_demo/5_CosMx/output #output directory
cluster_resolution: 0.3 #resolution for the FindClusters step, default 0.3
reference: humancortexref #Azimuth reference name, optional
reference_name: subclass #column name of the cell type label you would like to transfer. Required when reference is used.
integrate_data: True #True or False to merge/integrate all the data in the sampleMeta file
integrate_with_harmony: True #True or False to use Harmony for integration. Default as TrueThe sampleMeta.csv file contains sample name and several key input files of CosMx:
Sample,Directory,exprMat_file,fov_file,metadata_file,tx_file,polygon_file
S3,~/SpaceSequest_demo/5_CosMx/data/S3,S3_exprMat_file.csv,S3_fov_positions_file.csv,S3_metadata_file.csv,S3_tx_file.csv,S3-polygons.csv
S4,~/SpaceSequest_demo/5_CosMx/data/S4,S4_exprMat_file.csv,S4_fov_positions_file.csv,S4_metadata_file.csv,S4_tx_file.csv,S4-polygons.csvThen run the pipeline as below:
#Run the data
cosmx ~/SpaceSequest_demo/5_CosMx/config.yml8.3 Results
Finally check the results in the output_dir folder specified in the config.yml file. The output files contain several figures (UMAP for original cluster and annotated cell types), and Rdata files storing the results.
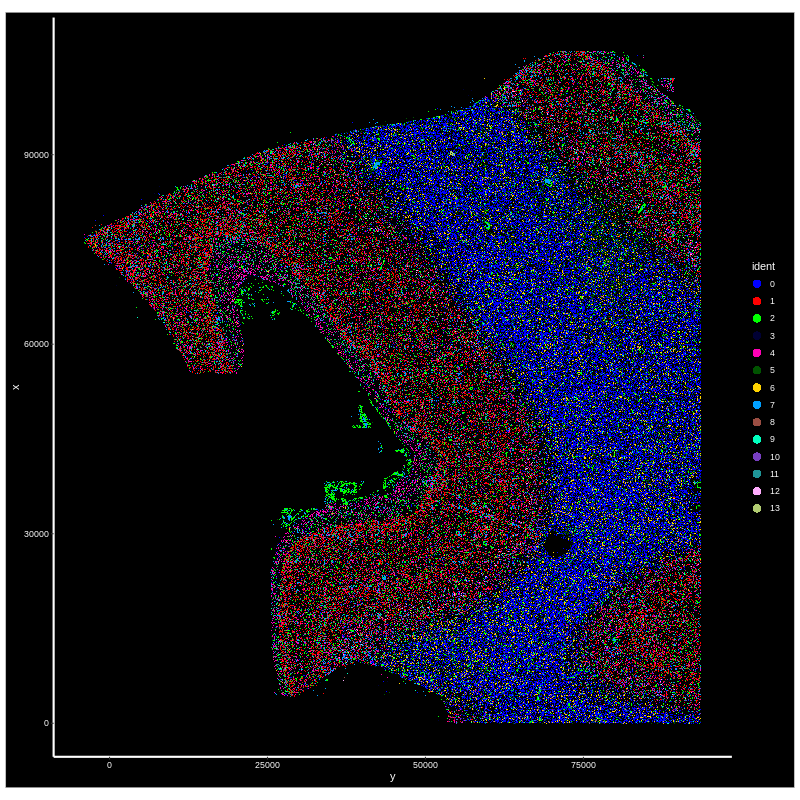
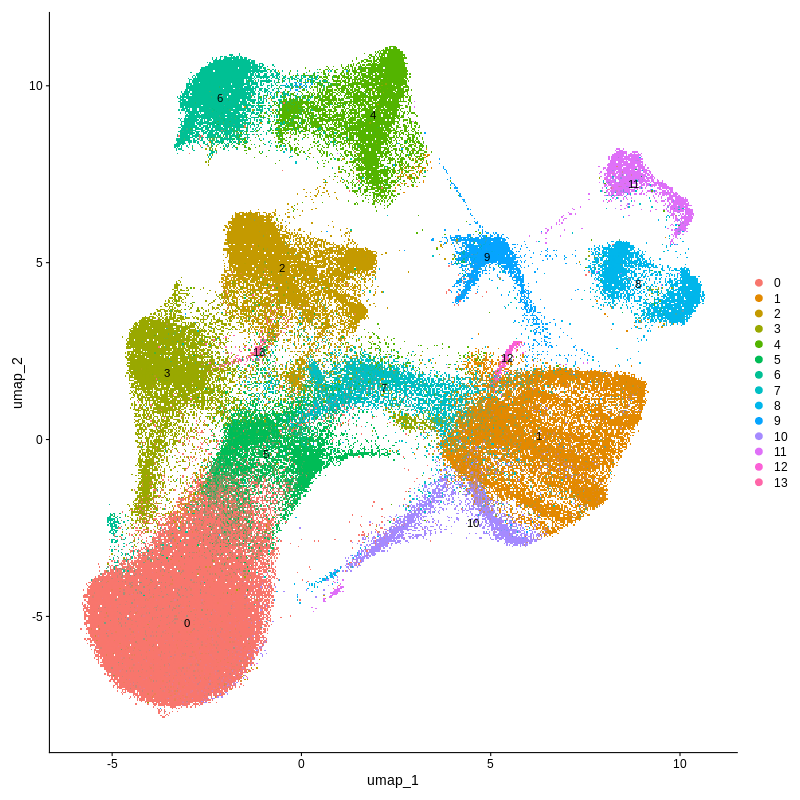
Figure 4. CosMX spatial-transcriptomics output. (Left) Tissue image with capture spots overlaid. (Right) UMAP embedding of spatial barcodes colored by cluster.