Chapter 11 Additional tools
11.1 scRMambient
scRMambient removes ambient RNA using the raw h5 file (raw_feature_bc_matrix.h5) as input, run CellBender, and output new h5 files for downstream analysis.
11.1.1 Set up the inputs
Besides the raw h5 file (raw_feature_bc_matrix.h5), the only required file for scRMambient is a sampleMeta_raw.csv file. There are several columns that are required: h5path, expected_cells, droplets_included, low_count_threshold,learning_rate. You can also add more sample metadata information, and they will be carried to a new sampleMeta.csv file generated after running scRMambient. Users can use that new sampleMeta.csv (it also contains some information related to CellBender run) to perform downstream scAnalyzer run.
expected_cells: This is the number of cells output by the Cell Ranger pipeline. We directly use the number here.
droplets_included: This is a critical parameter for CellBender. Please follow the instruction here to set it. We found that in many cases 15000 is a good number, but it also depends on your data.
low_count_threshold: We set to 15 by default.
learning_rate: We set to 0.0001 by default.
File hierarchy of the inputs:
~/test_ambient_rna_removal
├── Data1.raw_feature_bc_matrix.h5
├── Data2.raw_feature_bc_matrix.h5
└── sampleMeta_raw.csvAn example of the sampleMeta_raw.csv file:
Sample_Name,h5path,expected_cells,droplets_included,low_count_threshold,learning_rate
Data1,~/test_ambient_rna_removal/Data1.raw_feature_bc_matrix.h5,8119,15000,15,0.0001
Data2,~/test_ambient_rna_removal/Data2.raw_feature_bc_matrix.h5,7348,15000,15,0.000111.1.2 Run scRMambient
Then we run the pipeline:
scRMambient ~/test_ambient_rna_removal/sampleMeta_raw.csvThe output will be a new directory called cellbender, with new h5 files generated under the h5/ directory.
The file hierarchy is:
~/test_ambient_rna_removal
├── cellbender
├── all_cellbender.pdf # Plots generated by CellBender
├── all.log
├── cellbender_QC.pdf # QC plots
├── cellbender_rmRate.csv # Metrics related to the final cell number, and cell removal rates
├── h5/ # Directory for all output h5 files with ambient RNA removed
├── Data1.cellbender_cell_barcodes.csv
├── Data1.cellbender_filtered.h5
├── Data1.cellbender_filtered_qc.csv
├── Data1.cellbender.h5
├── Data1.cellbender.log
├── Data1.cellbender.pdf
├── ...
├── sc20230317_0 # File directory prepared for running scAnalyzer
├── config.yml
├── DEGinfo.csv
└── sampleMeta.csv # In this file, 'h5path' points to all new h5 files
├── Data1.raw_feature_bc_matrix.h5
├── Data2.raw_feature_bc_matrix.h5
└── sampleMeta_raw.csv11.2 scDEG
scDEG is a standalone pipeline to run the differential expression (DE) analysis independently if you alreadly have integrated scRNA-seq data in h5ad format.
scDEG uses the same methods as in the scRNASequest pipeline to run DE analysis. It just provides a flexible way to apply the same analysis outside the pipeline.
11.2.1 Initialize scDEG
scDEG requires three files:
- UMI: Gene counts matrix, can be in rds or h5ad format.
- Meta: cell type annotation and related meta information for DE analysis, in h5ad format. If you have this information in your h5ad matrix already, you can provide the same file twice (as UMI and meta file).
- config.yml: file storing the above UMI file path and meta data path, as well as the output file path.
By running the scDEG without providing any parameters, we can see the manual page of the script:
$ scDEG
scDEG /path/to/a/folder === or === scDEG /path/to/a/config/file
An empty config file will be generated automatically when a folder is provided
Powered by the Research Data Sciences Group [zhengyu.ouyang@biogen.com;yuhenry.sun@biogen.com]
------------Then providing a directory to scDEG, and it will generate a template config file:
$ scDEG /path/to/a/folder
#Example:
scDEG ~/DEG_resultsTemplate configuration file:
UMI: /path/to/Prefix.h5ad #required, can be a matrix rds or a h5ad file
meta: /path/to/Prefix.h5ad #required, can be a cell annotation data.frame rds or a h5ad file
output: /path/to/output/directory
DBname: cellxgeneVIP
parallel: slurm # False or "sge" or "slurm"
## DEG analysis for an annotation (such as disease vs health) within a cell type annotation
DEG_desp: /path/to/DEGinfo.csv #required for DEG analysis
# Please be causion of changing the following default filtering
# More details can be found: section 2.4 in https://pubmed.ncbi.nlm.nih.gov/35743881/
# Applies the 1st round of biostats filtering pipeline. Note that this filter is applied to all cells of the experiment
min.cells.per.gene: 3 # if `perc_filter` is FALSE, then keep only genes that have expression in at least min.cells.per.gene
min.genes.per.cell: 250 # keep cells with expression in at least min.genes.per.cell genes.
min.perc.cells.per.gene: 0.00 #if 'perc_filter'`' is TRUE, then keep only genes that have expression in at least min.per.cells.per.gene * 100 percent of cells
perc_filter: TRUE #if TRUE, apply the cells.per.gene filter using percentages (expressed as a decimal) rather than an absolute threshold
# Apply the 2nd round of biostats filtering. For "group" mode, the filtering is applied to `ref_group` and `alt_group` for the given cell type of interest.
R6_min.cells.per.gene: 3 #minimum cells expressed per gene. This filter is applied if `R6_perc.cells.filter` is FALSE
R6_min.perc.cells.per.gene: 0.1 # minimum % cells expressed per gene filtering (use decimal form of percentage). This threshold is applied if 'R6_perc.cells.filter' is TRUE and
'R6_cells.per.gene.filter' is TRUE
R6_min.cells.per.gene.type: "or" #The type of cell per gene filtering. If it has the value "and" then it requires the gene be expressed in both reference and non-reference grou
ps. If it has the value "or" then it requires the gene be expressed in either group
R6_cells.per.gene.filter: TRUE #TRUE means apply cells per gene filtering
R6_perc.cells.filter: TRUE #TRUE means apply cell.per.gene filtering by use of a percentage rather than absolute threshold. If the percentage results in a number less than R6_m
in.cells.per.gene, the code will automatically switch to min.cells.per.gene absolute thresholding
R6_perc.filter: FALSE #If TRUE, then apply the 75th percentile gene filtering
R6_perc.filter.type: "and" #The type of percentile gene filtering. If it has the value "and" then any gene that has 75th percentile of zero in both groups will be filtered out.
If it has the value "or" then any gene that has a 75th percentile of zero in either group will be filtered out.
R6_perc_threshold: 0.90 #Percentile threshold, 75th percentile is default. Express percentile as a decimal value.
R6_min.ave.pseudo.bulk.cpm: 1 #cpm filtering threshold
R6_pseudo.bulk.cpm.filter: FALSE #if TRUE, then apply a cpm filter on the pseudo-bulk counts
R6_min.cells.per.subj: 3 #Minimum cells required per subject, must be a nonzero numberThe UMI file can be a matrix rds or a h5ad file. The meta file can be a data.frame rds or a h5ad file, containing critical information that can be used for DE analysis.
## The config file for scDEG
UMI: ~/DEG_results/TST12055_raw_added.h5ad #required, can be a matrix rds or a h5ad file
meta: ~/DEG_results/TST12055_raw_added.h5ad #required, can be a cell annotation data.frame rds or a h5ad file
output: ~/DEG_results/
DBname: cellxgeneVIP
## DEG analysis for an annotation (such as disease vs. health) within a cell type annotation
DEG_desp: ~/DEG_results/DEGinfo.csv #required for DEG analysis
## Please be cautious of changing the following default filtering.
min.cells.per.gene: 3
min.genes.per.cell: 250
min.perc.cells.per.gene: 0.00
perc_filter: TRUE
## ...11.2.2 Run DE analysis
Please refer the previous section Differential expression (DE) analysis to prepare the DE comparison file.
After preparing the config.yml and DEGinfo.csv files, run the pipeline:
$ scDEG /path/to/config.yml
#Example:
scDEG ~/DEG_results/config.yml11.3 scTool
We also provide scTool, a user-friendly toolkit for modifying the h5ad file. With scTool, the user can:
Remove existing annotations in the data, such as the cell type annotation.
Add cell type annotation by providing a two-column csv file, specifying the cell type category of each single cell.
Export a list of genes along with all annotation.
By running the scTool without providing any parameters, we can see the manual page of the script:
$ scTool
usage: scTool.py [-h] {rm,add,export,pseudo} h5ad [changes]
Additional sc tools. WARNING: The input h5ad files will be over-written!
positional arguments:
{rm,add,export,pseudo}
Modify the h5ad by either "rm", "add" or "export" cell level annotations.
Create pseudo bulk as RNAsequest analysis input by "pseudo"
h5ad Path to a h5ad file to be modified or extract from (raw UMI for pseudo)
changes Options:
1. (rm) A list of annotaions to be removed (separated by ",")
2. (add) A path to a csv file contains cell level annotations (first column is the cell ID)
3. (export) A list of genes (separated by ",", empty or max 500) to be exported along with all annotations.
4. (pseudo) A list of obs to group cells into pseudo bulk, separated by ","
optional arguments:
-h, --help show this help message and exitWe use this pubic E-MTAB-11115 data as an example:
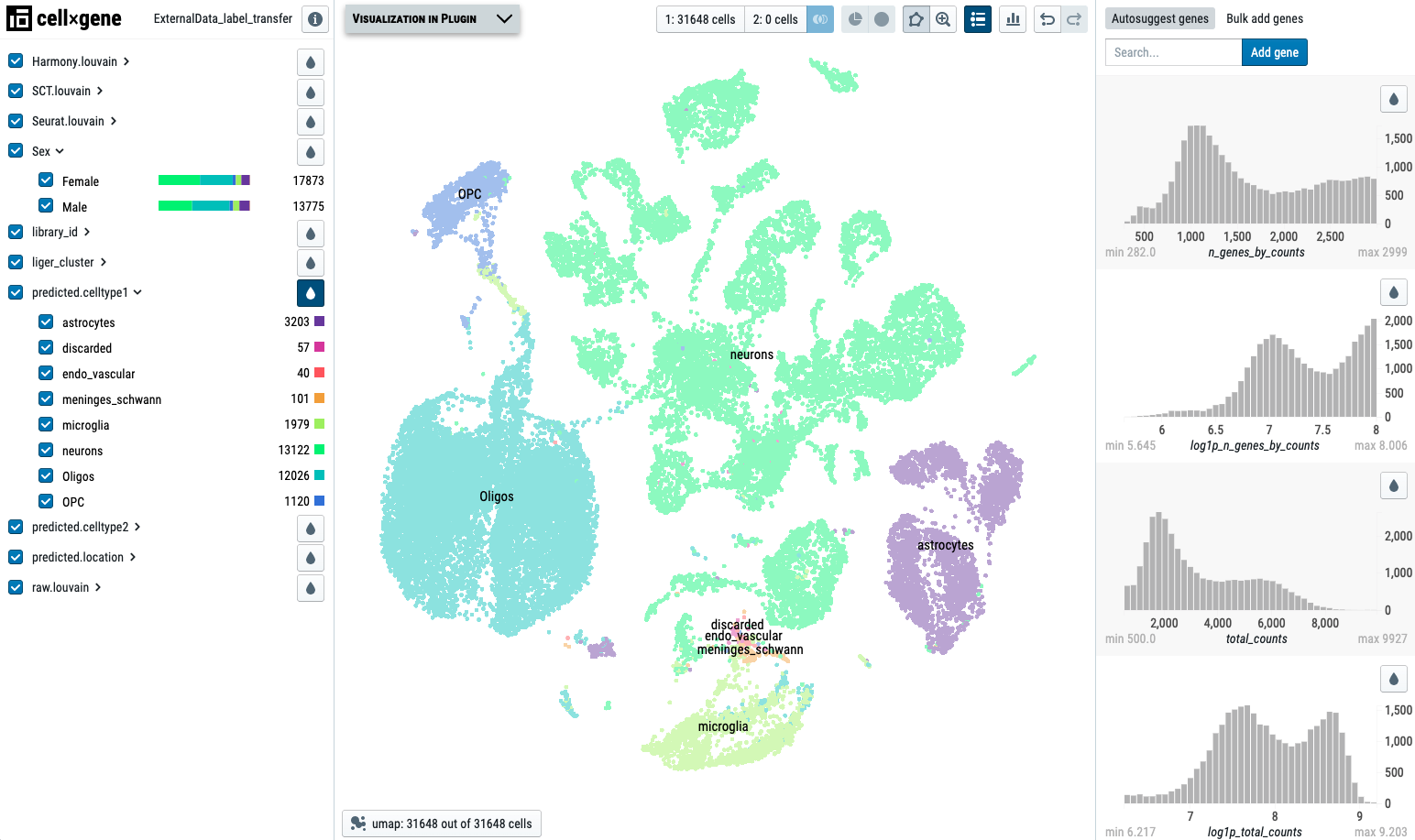
11.3.1 Remove annotations
To remove annotation, we can simply pass the annotation header names to the program, and it will remove these headers, and re-write the original file. Please back-up the original file first if you would like to keep a copy ot it. As an example, here we remove the following headers: predicted.celltype2 and predicted.location.
scTool rm ProjName_raw_added.h5ad predicted.celltype2,predicted.location11.3.2 Add annotations
To provide a new annotation, the user needs to prepare a csv file containing cell barcode and new annotation names. For example, we use the following csv file to include a manual.curated.labels cell type labeling to the original data:
$ head -2 manual.curated.labels.csv
AAACCCAAGGAAGTAG-1-5705STDY8058280,neurons
AAACCCAAGGGCAGTT-1-5705STDY8058280,Oligos
#Run the scTool:
scTool add ProjName_raw_added.h5ad manual.curated.labels.csv11.3.3 Export gene expressions
This function exports gene expression values of provided genes, along with all other annotations. This provides a flexible way to examine the expression values of a set of genes.
scTool export ProjName_raw_added.h5ad App,Olig1,P2ry12The output result is a .csv file: ProjName_raw_added.h5ad.csv. Here is an example of the output file:

11.3.4 Pseudo-bulk aggregation
The “scTool pseudo …” will create a folder “pseudoBulk” where the h5ad is.
scTool pseudo ProjName_raw_added.h5ad orig.ident,cell_typeOutput: Four files will be generated in the folder:
genes.UMI.tsv: the UMI (raw counts) for pseudo bulk samples (columns)
samplesheet.tsv: contains obs (if the unique value is only one for each of pseudo bulk)
genes.CPM.tsv: contains normalized expression (count per million)
genes.CPKcell.tsv: contains normalized express (count per 1k cells)
The output can be used in our RNASequest pipeline: https://github.com/interactivereport/RNASequest
In the RNASequest pipeline, EAinit will treat this as external project, and user will have to fill in:
proj_name, prj_counts (genes.UMI.tsv), prj_TPM(genes.CPM.tsv or genes.CPKcell.tsv), sample_meta (samplesheet.tsv), speciesAlso, in RNASequest, prj_effLength is not required, but covariates_adjust will be disabled, and gene_annotation is not required. The first column in count matrix will be used as gene name.
11.3.5 Demo scTool
We use the h5ad file generated from demo data set to test scTool: scRNASequest_demo.h5ad.
scTool export scRNASequest_demo.h5ad App,Olig1,P2ry12The output will be a scRNASequest_demo.h5ad.csv file.